


本站讯 近日,中国海洋大学医药学院、海洋药物教育部重点实验室任为武教授课题组在多环西松烷降二萜海洋天然产物全合成方面取得新进展,相关研究成果以“Asymmetric Total Synthesis of (+)-Ineleganolide”((+)-Ineleganolide的不对称全合成)为题,发表在国际知名学术期刊Angewandte Chemie International Edition(《德国应用化学》)。
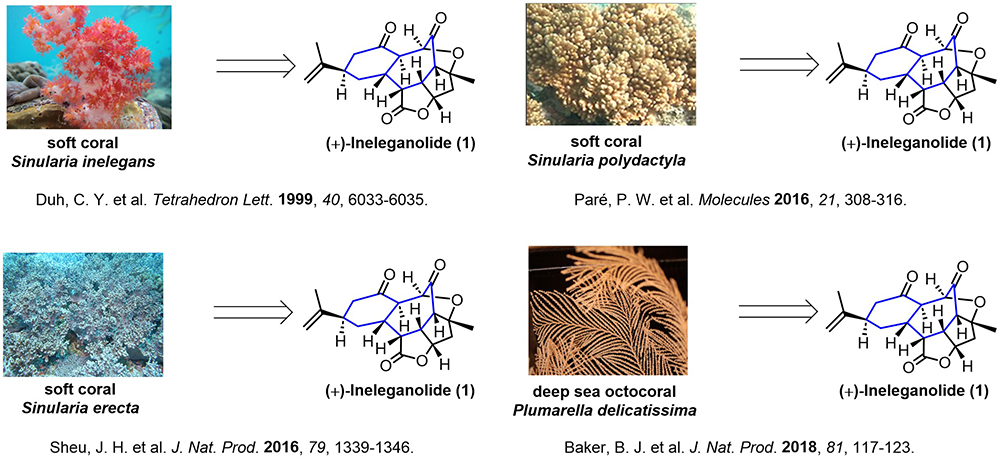
图1 Ineleganolide的分离工作报道
天然产物结构的复杂性、多样性与生物活性的特异性,共同赋予其独特的研究价值,成为推动药物研发、合成策略创新及生命科学研究的核心驱动力。多环西松烷二萜与降二萜是一类重要的海洋天然产物,软珊瑚为其最主要的生物来源。该类分子普遍具有高度氧化的复杂稠环骨架,且大多展现出优异的抗肿瘤药理活性,近三十年来一直是合成领域的前沿研究方向。Ineleganolide是此类天然产物中最具代表性的分子之一,于1999年从软珊瑚Sinularia inelegans中分离得到,随后陆续有数个课题组完成对其分离工作(图1)。生物活性实验证实,ineleganolide对小鼠白血病细胞系P-388具有显著的细胞毒作用。从结构层面来看,该分子拥有刚性强、环张力突出的 [6,7,5,5,5] 五环笼型骨架,分子内共包含9个手性中心,其中8个为连续的手性中心,立体化学控制困难,在合成上极具挑战性。
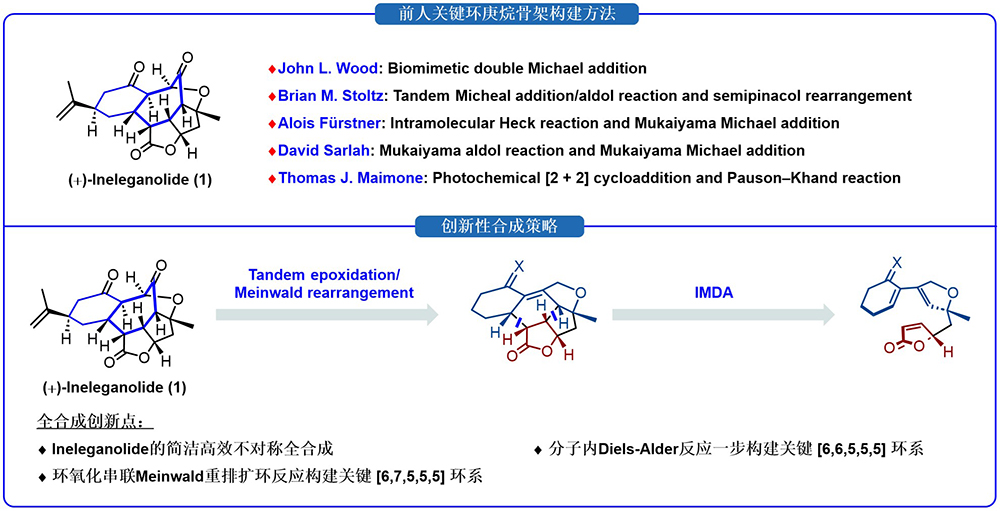
图2 (+)-Ineleganolide的创新性合成策略
针对该分子独特的结构,研究团队提出了全新的不对称合成策略:先借助分子内Diels–Alder反应一步构筑 [6,6,6,5,5] 稠合五环母核,再利用环己烷骨架固有环张力驱动环氧化串联Meinwald重排反应实现扩环,高效构建了目标分子中拥挤的环庚烷骨架(图2)。依托该创新合成策略,最终团队仅通过13步线性反应,简洁高效地完成了 (+)-ineleganolide的不对称全合成。
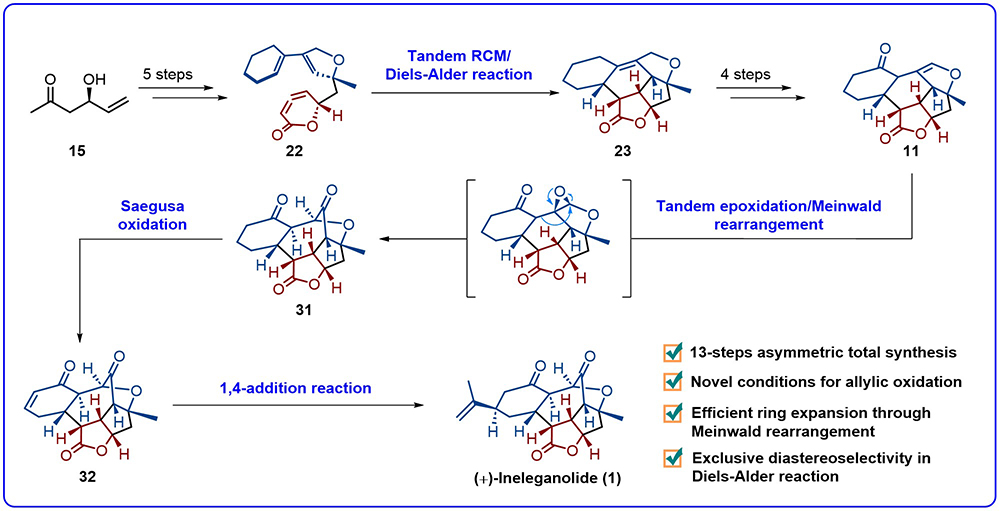
图3 (+)-Ineleganolide的不对称合成路线
研究团队从商品化手性原料15出发(图3),经一锅法炔丙基锌试剂加成串联选择性TBS保护反应、金催化环化反应、引入OTf基团、Suzuki偶联反应以及酯缩合反应等5步反应,完成对关键底物前体22的制备,随后经RCM串联分子内Diels–Alder反应,完成对 [6,6,6,5,5] 稠合五环母核的构建得到中间体23。通过一锅法Riley氧化串联Ley氧化反应、选择性烯丙位氧化反应、还原反应以及消除等4步反应完成了前体10的构建,而后经过环氧化串联Meinwald重排反应制备关键底物31,最后经Saegusa氧化反应及1,4加成反应,成功完成对 (+)-ineleganolide的不对称全合成。本合成策略步骤精简高效,且仅包含13步线性转化,合成过程中的RCM串联分子内Diels–Alder环加成与环氧化串联Meinwald重排反应,均生成单一构型产物。该合成路线不仅实现目标分子的高效制备,也为西松烷降二萜天然产物家族的系统成药性研究提供重要物质基础。

团队合影(前排右三为任为武教授)
中国海洋大学为该研究成果的第一通讯单位,医药学院任为武教授为通讯作者,医药学院韩长宏博士为第一作者。研究工作得到了国家自然科学基金、泰山学者青年专家等项目支持。
编辑:赵奚赟
责任编辑:刘莅










































