


本站讯 近日,中国海洋大学医药学院、海洋药物教育部重点实验室王洪玉教授联合山东师范大学刘建标副教授,在环丁烷开环双官能化构建结构多样性烷烃类化合物方面取得新进展。相关研究成果以“Nickel-Catalyzed Ring-Opening Difunctionalization of Methylenecyclobutanes via Strain-Release-Enabled C-C Activation”(镍催化亚甲基环丁烷开环双官能化)为题,发表在国际知名学术期刊Nature Communications(《自然·通讯》)上。
从简单模块高效组装复杂分子结构是催化领域的重要目标之一,这对于合成化学、药物发现以及材料科学均具有深远意义。在这一背景下,能够在温和条件下通过一步反应同时构建多个碳–碳键的催化多组分反应尤为重要,不仅提高了原子经济性并减少了废物产生,还能简化合成路线。该领域的重要突破之一是末端烯烃和炔烃的1,2-双官能化,底物来源广泛且易于获取,该类反应在过渡金属催化、光催化、电催化以及协同催化体系的推动下取得了显著进展。然而,这些方法通常仅生成两个C(sp³)或两个C(sp²)杂化碳中心,从而限制了对不同杂化类型碳骨架分子的获取。经一步反应同时构建C(sp³)和C(sp²)中心仍然是一个关键挑战,末端联烯在此方面表现出独特优势,其正交π体系和独特电子性质为解决该问题提供了可能。通过区域选择性和化学选择性的双官能化,联烯能够同时构建C(sp³)/C(sp²)碳中心,为获得高度官能化的三维结构提供更直接的高效路径。
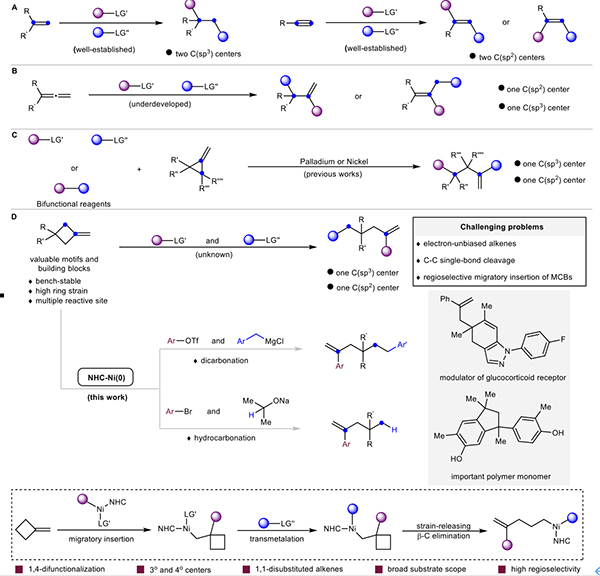
图1 Ni-NHC催化亚甲基环丁烷双官能化
亚甲基环丁烷(MCBs)作为是一类多功能有机试剂,在化学合成中具有重要潜力。其分子中同时蕴含较高的环张力储能(环丁烷:26.5 kcal/mol,环丙烷:27.5 kcal/mol)以及C=Cπ体系与环丁烷σ体系所赋予的双重反应性。这种特性使其既可通过C=C键双官能化发生转化,也可通过应变驱动的C–C键断裂实现结构重组,从而显著拓展MCBs的化学空间。目前,关于MCBs应用的研究已有较多报道。然而,现有研究主要集中在C=C键的1,2-双官能化以及环扩张反应,而通过选择性C–C键断裂实现环开裂并进行1,4-双官能化的策略仍相对匮乏,这在一定程度上限制了其在多组分复杂分子构建中的应用潜力。
鉴于亚甲基环丁烷中C=C键具有较高环张力且电子性质相对中性,实现其区域选择性的迁移插入既是基础科学问题,也是实现高效开环官能化的关键前提。研究团队发展了Ni–NHC催化体系,克服了MCBs内在的空间位阻和电子效应偏向,通过对亚甲基的支链选择性插入,引发应变驱动的β-碳消除,从而实现C–C键的直接断裂及后续的1,4-双官能化过程,实现了亚甲基环丁烷的高效1,4-二碳官能化和1,4-氢碳官能化反应,该策略能够构建非相邻的sp²与sp³碳中心,这些结构单元在高附加值分子的构建中具有重要意义。

团队合影(前排中为王洪玉教授)
中国海洋大学为本文的第一通讯单位,中国海洋大学医药学院王洪玉教授与山东师范大学刘建标副教授为共同通讯作者,中国海洋大学医药学院硕士生王婷婷、刘海娜、罗美虹以及山东师范大学硕士生赵雯雯为共同第一作者。研究工作得到了国家自然科学基金、山东省自然科学基金、泰山学者青年专家、中国海洋大学青年英才启动经费等项目资助。
文章链接:https://doi.org/10.1038/s41467-026-71557-y
编辑:赵奚赟
责任编辑:刘莅










































