


本站讯 一个完整且注释精准的基因组是解析生物学功能和调控机制的核心基石。基因注释包括编码蛋白基因的编码区(CDS)、非编码区(UTR)、转录起始位点(TSS)和转录终止位点(TES)、非编码RNA,以及基因的可变剪切亚型等关键特征的识别的标注。只有通过高质量的基因注释,才能将基因组序列转化为具有生物学意义的信息。

嗜热四膜虫(Tetrahymena thermophila)是一种广泛应用于基础生物学研究的单细胞模式真核生物,曾因核酶和端粒酶的研究分别斩获1989年诺贝尔化学奖和2009年诺贝尔生理或医学奖。四膜虫大核基因组已历经多次组装和注释,其中包括2020年由学校海洋生物多样性与进化研究所原生动物学团队高珊教授课题组率先完成的端粒到端粒的完整基因组组装。然而,现有的基因注释仍存在诸多不足,例如基因错误注释、TSS和TES位置不准确,以及UTR信息缺失等。近日,高珊教授课题组通过整合大规模的转录组数据和表观遗传学数据,运用机器学习模型、人工核对和实验验证,深度优化了嗜热四膜虫大核基因组的注释,相关成果Comprehensive genomeannotation of the model ciliate Tetrahymena thermophila by in-depth epigenetic and transcriptomic profiling(利用大规模的转录组和表观组数据深度优化模式生物嗜热四膜虫的基因组注释)于2024年12月9日在线发表于Nucleic Acids Research杂志上。
研究团队收集了四膜虫不同生长阶段(生长、饥饿和接合生殖)的大规模RNA-seq数据,结合纳米孔直接RNA测序(Nanopore DRS)、链特异性RNA-seq等技术,对基因模型进行了全面校正和重注释。与此同时,还引入了H3K4me3、H2A.Z、6mA、核小体等表观遗传标记,使用机器学习算法进一步优化了基因注释。通过对表观组和转录组数据的整合(图1),团队成功预测了24351个TSS,并基于Cap-seq数据验证了这些TSS的准确性。最终,优化后的基因组新增了2481个基因,并对23936个基因模型进行了修订,其中包括外显子改变、基因合并、基因拆分以及基因方向反转等。此外,研究团队首次注释了26047个基因的UTR,并鉴定出5500个基因的8,339种可变剪切亚型。这些优化显著提升了四膜虫基因组注释的完整性和准确性,不仅强化了四膜虫作为遗传工具在生物学研究中的实用价值,还为其他真核生物的基因组注释提供了有益的借鉴。
团队同时鉴定了5525个天然反义转录本(NATs),并发现约20%的蛋白编码基因存在反义转录。NATs通常较短且低表达,但在四膜虫的有性(接合)生殖阶段,其表达水平显著升高。进一步分析表明,大部分NATs与其正义蛋白编码基因呈现互斥的时间特异性表达模式,可能通过与正义基因相互作用,调控其转录或翻译。这一发现为揭示四膜虫的转录调控机制提供了新的视角。
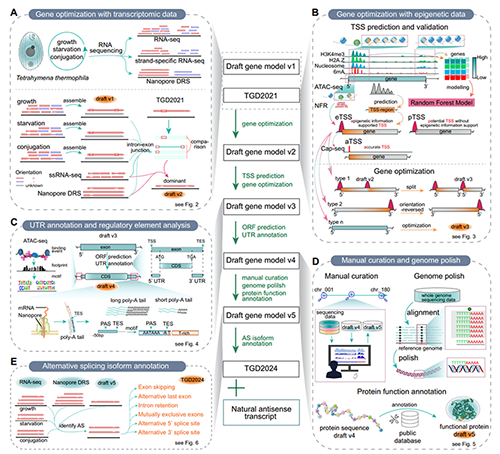
整合转录组和表观遗传数据优化基因模型示意图
学校海洋生物多样性与进化研究所原生动物学团队高珊教授为文章的通讯作者。博士生叶飞、山东大学陈晓教授和博士毕业生李源为共同第一作者。硕士生鞠艾利、博士毕业生盛亚岚、博士生段丽丽、张佳晨和张喆,以及美国布拉德利大学Naomi A. Stover副教授等对本文亦有重要贡献。该工作得到学校海洋生物多样性与进化研究所超算集群、崂山实验室高性能科学计算与系统仿真平台和中国海洋大学高等海洋研究院海洋大数据中心等计算资源的资助。
通讯员: 张川
原文链接:https://academic.oup.com/nar/advance-article/doi/10.1093/nar/gkae1177/7919455
编辑:赵奚赟
责任编辑:刘莅










































