


本站讯 10月5日,美国化学会旗舰会刊Journal of the American Chemical Society在线发表了中国海洋大学功能分子合成与应用研究中心、海洋药物教育部重点实验室徐涛课题组关于海洋来源抗疟天然产物全合成的最新研究成果《天然产物(−)-Caulamidine D和(−)-Isocaulamidine D的首次不对称全合成及其绝对构型修订》(Enantioselective Total Syntheses of (−)-Caulamidine D and (−)-Isocaulamidine D and Their Absolute Configuration Reassignment)。
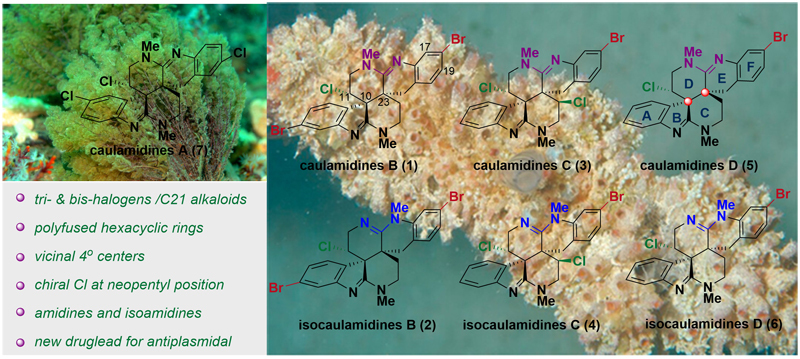
图1. 深海苔藓虫和海鞘中分离获得Caulamidines A & Isocaulamidines B~D
海洋生物对于海水中卤素(Cl, Br, I)的利用是其区别于陆生生物的“特殊技能”。含有卤素“点缀”的天然分子,特别是含Br和I的天然产物是其海洋来源的重要特征。卤素是海洋生物用来“调整”其多用途次级代谢产物的一种独特的策略,通常会导致次级代谢产物生物活性的改变。引入卤素不可避免地增加了天然产物结构的复杂性和化学鉴定的难度。多卤化生物碱Caulamidines B-D和Isocaulamidines B-D是由Gustafson小组从帕劳群岛附近的海鞘Polyandrocarpa sp.中分离得到的系列含Cl和Br的生物碱。研究表明,这些天然产物在NCI-60细胞筛选中表现出中等细胞毒性,其同系物Caulamidines A和B则表现出了对于氯喹耐药型疟原虫的抑制活性,其前所未有的分子结构和优良的生物活性,使(iso)caulamidines类化合物成为潜在的抗疟原虫药物先导化合物,引起了化学合成和新药研发领域科研人员的广泛关注。
徐涛课题组以简单的原料色醇衍生物为初始原料,经过一步不对称的Meerwein-Eschenmoser-Clasien重排反应(MECR)获得关键中间体,并通过Red-Al介导的环化关环构建A-B-C并环体系。接下来将侧链OTIPS转化为醛基,并在醛基α位引入手性氯原子,得到串联环化的前体。随后通过铁粉氯化铵的条件进行硝基还原,同时进行6-exo-dig/6-exo-tet氨基/氰基串联环化反应一步构建上侧D-E-F并环环系。从而成功构建出6/5/6/6/6/6六环骨架,最后在碘甲烷的作用下,顺利得到海洋天然产物(−)-Caulamidine D和(−)-Isocaulamidine D。
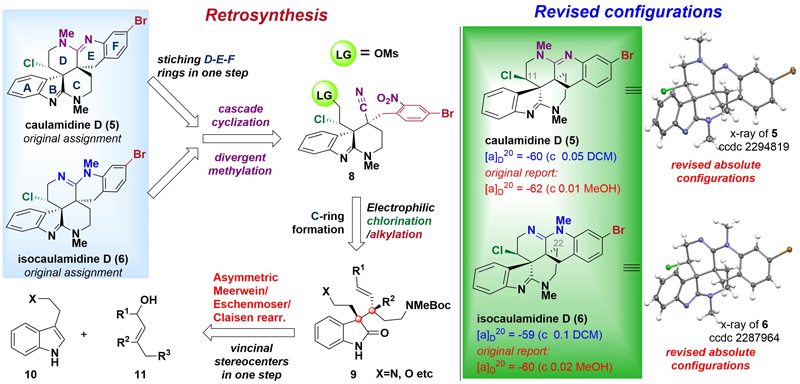
图2. Caulamidine D和Isocaulamidine D的逆合成分析与结构修订
研究成果首次完成了(−)-Caulamidine D及其同系物(−)-Isocaulamidine D的对映选择性全合成,并基于X射线晶体学检测重新确定了它们的绝对构型。在核磁共振数据对比时发现,天然样品的Caulamidine D和Isocaulamidine D实际上是TFA盐的形式存在。通过徐涛课题组开发的统一的合成策略,仅需16步(最长线性步骤)即可同时合成天然产物5和6。关键反应包括:开发和应用不对称Meerwein-Eschenmoser-Claisen重排,构建键C10、C23连续立体中心;提出并实践了6-exo-dig/6-exo-tet胺基/氰基串联环化策略。值得强调的是,徐涛教授开发的反应路线具有可规模化制备的巨大优势,为该家族天然产物的大量制备提供了高效途径,这也是基于化学合成解决海洋药物“药源”瓶颈的典型例子之一。
研究成果的第一完成单位是中国海洋大学医药学院,通讯作者徐涛教授是中国海洋大学“筑峰”三层次教授,是国家优青和山东省杰出青年基金获得者、首批泰山学者青年专家和海洋国家实验室“鳌山人才”优秀青年学者。文章第一作者是医药学院博士生于海勇。该工作得到了国家自然科学基金项目,以及山东省杰出青年基金和学校杰青培育计划等经费的大力支持。
编辑:刘莅
责任编辑:赵奚赟










































